CLSCC Chloride Stress Corrosion Cracking Mechanism
The mechanism of Chloride Stress Corrosion Cracking CLSCC is complex and the current understanding is discussed in Section 5 of this report. Essentially CLSCC involves a combination of the electrochemistry of metal dissolving over a highly localised area, i.e. at the base of a pit or crevice, and microstructural processes that separate the metal structure in a region of highly localised plastic strain, i.e. at the crack tip. A detailed review of candidate mechanisms is set out in the literature survey. For the purposes of this report, the mechanism will be described simply in terms of an initiation stage, dominated by electrochemical mechanisms, and a crack propagation stage in which both electrochemistry and metal separation are involved.
The high corrosion resistance of austenitic stainless steel in most atmospheric and aqueous
environments is due to passivation by a thin (~2nm) layer of chromium oxide. Wet and humid environments containing chloride ions can cause pitting corrosion and crevice corrosion of austenitic stainless steel components. Components under an applied or residual stress can deteriorate further by stress corrosion cracking in these conditions. Pitting is simply a breakdown of the chromium oxide layer followed by localised corrosion that produces pits, which may cause perforation of a vessel or pipework.
Pitting is mainly associated with
microscopic heterogeneities in a surface rather than macroscopic physical features of a
component. Crevice corrosion is also a breakdown of the chromium oxide layer followed by
localised corrosion but in contrast to pitting, it occurs at specific physical features where a
surface is partly shielded and stagnant solution exists at an interface with the shielded area.
Where pitting or crevice corrosion causes localised metal loss, the geometry and the local
environment are critical in ensuring that metal loss becomes self-sustaining.
Localised
corrosion in an active pit or inside a crevice produces a solution with the following
characteristics.
(i) Higher chloride level than the bulk solution because negatively charged chloride
ions migrate into the pit to balance the positively charged metal ions.
(ii) Strongly acidic with a very low pH (~0).
(iii) Nearly saturated with complex ions produced from dissolved metal, chloride and water.
This demonstrates an essential feature of localised corrosion on stainless steel, i.e. the region
inside a pit, a crevice or a crack is an isolated electrochemical cell that contains a much more
aggressive environment than the bulk solution.
It is commonly accepted that CLSCC initiates from sites of active pitting or crevice corrosion and therefore, cracks are considered to grow in the high chloride, strongly acidic, nearsaturated solution that develops at sites of localised corrosion. One theory proposes that CLSCC only occurs when a crack grows more quickly than the rate of metal removal by localised corrosion from the base of a crevice or pit; in other words there is a competition
between the rate of CLSCC and the rate of localised corrosion. Crack growth is also restricted
to a range of electrochemical potential that is defined by an upper limit where dissolution
exceeds crack growth and a lower limit set by re-passivation.
This approach has been used by
Tsujikawa to demonstrate a temperature dependency of CLSCC because crack growth increases
more rapidly with temperature than the rate of localised corrosion. Figure 1 is a schematic
description of Tsujkawa’s theory. In our opinion, the competition theory reinforces the fact
that localised corrosion is a prerequisite for CLSCC propagation in austenitic stainless steel.
Stress corrosion tests carried out by Tsujikawa and recent tests carried at HSL have
confirmed that it is very difficult to initiate CLSCC on bare, smooth specimens under laboratory
conditions when there is no localised corrosion of the surface under stress.
The susceptibility of austenitic stainless steels to CLSCC depends on a range of environmental
variables that include chloride concentration, temperature and pH. Other variables include, for
example, stress level, surface finish and the metallurgical condition of the steel. The approach
in this report is therefore based on assessing how variables are likely to affect:
(i) The initiation of CLSCC by localised corrosion.
(ii) Crack propagation when the rate of CLSCC exceeds the rate of localised
corrosion.
Figure 1. Schematic graph to demonstrate the competition concept proposed by
Tsujikawa. Note how changes in crack growth rate or corrosion rate can
alter the ‘critical’ temperature for CLSCC.
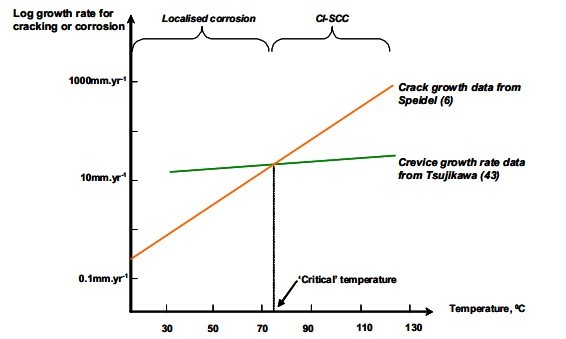
Note that Tsujikawa found that the ‘critical’ temperature could range from 42 C to 130 C by varying the phosphorus and copper levels in the steel.
Related References:
1. austenitic stainless steel
2. Stress Corrosion Cracking SCC
3. Chloride Stress Corrosion Cracking (CLSCC)
4. Stress Corrosin Cracking SCC of Duplex Stainless Steel
5. Chloride Stress Corrosion Cracking in Austenitic Stainless Steel
6. Recommendations for Assessing Susceptibility to CLSCC
7. Main Findings on CLSCC in the Reactors
8. Literature Review to Chloride Stress Corrosion Cracking
9. CLSCC Chloride Stress Corrosion Cracking Mechanism
10. Factors Affecting CLSCC Chloride Stress Corrosion Cracking
11. Controlling Chloride Stress Corrosion Cracking
12. Structural Integrity Assessment
13. Non-Destructive Examination NDE
|
